使用不同方法规一化、PCA和批次效应矫正的差异
目录
1)规一化表达矩阵
转录组差异表达分析时,需要对表达矩阵进行规一化。比较常用的方法有log2和DESeq2包的vst和rlog方法。那么它们有什么不一样呢?我们拿DESeq2的示例数据进行比较。
library(RNAseqFlow)
library(DESeq2)
library(corrplot)
input <- createCountPhe() # 得到表达矩阵和表型信息
expr <- input[[1]]
phe <- input[[2]]
dds <- create_DEseq(count_data=expr,col_data=phe,design_names = "condition+type",group_name ="condition",ref_level = "untreated") # 创建DESeqDataSet对象
## [1] "The id order between gene count file and phenotype file is identical without modification!"
dds
## class: DESeqDataSet
## dim: 10089 7
## metadata(1): version
## assays(4): counts mu H cooks
## rownames(10089): FBgn0000008 FBgn0000017 ... FBgn0261574 FBgn0261575
## rowData names(26): baseMean baseVar ... deviance maxCooks
## colnames(7): treated1 treated2 ... untreated3 untreated4
## colData names(3): condition type sizeFactor
norm <- log2(expr+1) #直接用log2进行规一化
norm[1:5,1:5]
## treated1 treated2 treated3 untreated1 untreated2
## FBgn0000008 7.139551 6.475733 6.149747 6.539159 7.339850
## FBgn0000017 12.599448 11.585432 11.703471 12.187661 13.089285
## FBgn0000018 9.497852 8.228819 8.271463 9.189825 9.573647
## FBgn0000024 3.459432 3.000000 2.584963 3.459432 3.584963
## FBgn0000032 10.730470 9.445015 9.566054 10.498849 10.743151
ntd <- normTransform(dds) # DESeq中的函数
assay(ntd)[1:5,1:5]
## treated1 treated2 treated3 untreated1 untreated2
## FBgn0000008 6.436242 6.865379 6.410556 6.354468 6.504519
## FBgn0000017 11.889798 11.978840 11.967612 12.000870 12.247040
## FBgn0000018 8.789322 8.621190 8.534895 9.003332 8.732772
## FBgn0000024 2.830673 3.349813 2.808365 3.290618 2.834907
## FBgn0000032 10.021212 9.838041 9.829949 10.312153 9.901443
normalized_counts = counts(dds, normalized=TRUE)
ntd1 = log2(normalized_counts+1)
ntd1[1:5,1:5]
## treated1 treated2 treated3 untreated1 untreated2
## FBgn0000008 6.436242 6.865379 6.410556 6.354468 6.504519
## FBgn0000017 11.889798 11.978840 11.967612 12.000870 12.247040
## FBgn0000018 8.789322 8.621190 8.534895 9.003332 8.732772
## FBgn0000024 2.830673 3.349813 2.808365 3.290618 2.834907
## FBgn0000032 10.021212 9.838041 9.829949 10.312153 9.901443
rld <- rlog(dds, blind=FALSE) # DESeq中的函数
assay(rld)[1:5,1:5]
## treated1 treated2 treated3 untreated1 untreated2
## FBgn0000008 6.499251 6.678569 6.494630 6.469361 6.527893
## FBgn0000017 11.933399 12.001811 11.993193 12.018453 12.208070
## FBgn0000018 8.758693 8.649608 8.594954 8.900817 8.721092
## FBgn0000024 2.679631 2.713855 2.679046 2.712598 2.679846
## FBgn0000032 9.998801 9.867264 9.861304 10.211745 9.911647
vsd <- vst(dds, blind=FALSE) # DESeq中的函数
assay(vsd)[1:5,1:5]
## treated1 treated2 treated3 untreated1 untreated2
## FBgn0000008 8.005828 8.192922 7.995328 7.972665 8.034115
## FBgn0000017 11.968753 12.053246 12.042577 12.074190 12.309219
## FBgn0000018 9.322821 9.204951 9.145817 9.477809 9.282788
## FBgn0000024 7.105652 7.182429 7.102606 7.173068 7.106233
## FBgn0000032 10.283202 10.130746 10.124080 10.531172 10.183179
sample <- cbind(log2=norm[,1],ntd=assay(ntd)[,1],rld=assay(rld)[,1],vsd=assay(vsd)[,1])
corr <- cor(sample,use="pairwise.complete.obs")
corrplot(corr, order = "original", type = "upper", mar=c(1,5,5,1),tl.pos="n")
corrplot(corr, add = TRUE, type = "lower", method = "number", order = "original",
tl.pos="full",cl.pos="n")
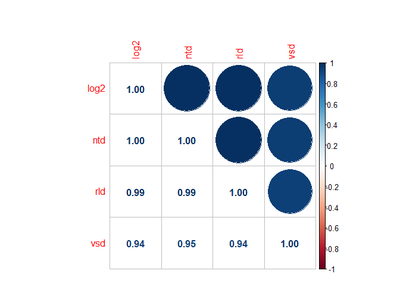
从数值来看,不同方法相差不大。从相关性来看,不同方法规一化的数据差异也不大,以第一个样本为例,相关性都达到0.94以上。因此,不同规一化方法差别不是特别大,选择其中一个就好。
不同方法的运行速度:
- log2 方法最简单,最方便。
- normTransform 与log2类似,但是它会先进行size factor的规一化再log2。需要首先转成DESeqDataSet类。
- rlog 速度比较慢。需要首先转成DESeqDataSet类。
- vst是rlog的快速版。需要首先转成DESeqDataSet类。
2)PCA 展示
如果输入数据是DESeqDataSet类,可以使用plotPCA进行展示:
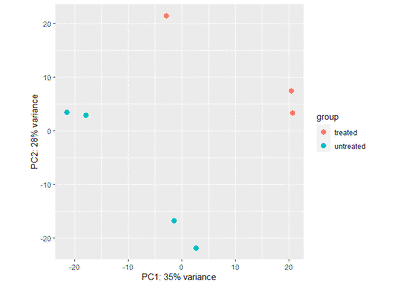
plotPCA(ntd,intgroup=c("condition"))
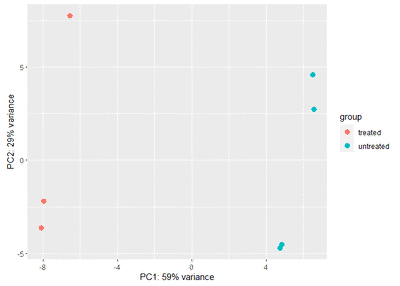
plotPCA(rld,intgroup=c("condition"))
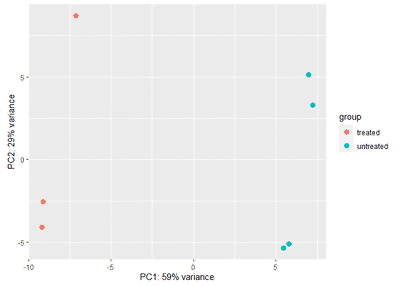
plotPCA(vsd,intgroup=c("condition"))
如果是log2的结果,可以用plot_PCA展示:
library(bioTools)
library(ggplot2)
plot_PCA(t(norm),phe=input[[2]],group = "condition")
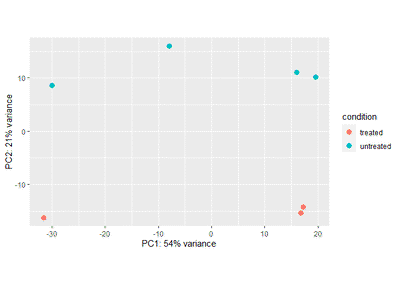
plot_PCA是bioTools包的函数,可用devtools::install_github("Feng-Zhang/bioTools")进行安装。
plot_PCA会使用prcomp函数计算距离矩阵,再用ggplot2画图。prcomp函数的输入文件是data.frame(n*m),行(n)为样本,列(m)为多维变量。prcomp会将多维变量降维到n维。
注意如果mat是没有规一化的数据,需要设置其中的参数scale=T,将数据进行规一化。
可以看到rlog和vst的结果是类似的,且比normTransform方法好,因为PC1和PC2加起来解释的方差更大。而log2方法的效果也很不错。因此:
- 如果使用DESeq2进行相关分析时,推荐使用vst方法。
- 如果输入文件是一般的表达矩阵,用log2的方法也是不错的选择,不用费劲地转成DESeqDataSet类。
- 用plot_PCA可以方便地画PCA图。它是用ggplot2画的,返回的结果还可以用ggplot2进一步优化。
3)查看批次效应
我们一般用PCA方法来看样本间是否有批次效应。 本例中condition是我们关注的因素,但是type不是,它是指不同测序平台类型。我们不希望不同平台的数据对我们所关注的因素造成影响,即不希望不同平台的测序结果有系统性差异。因此可以先用PCA方法查看type对样本的影响。
plotPCA(ntd,intgroup=c("type"))
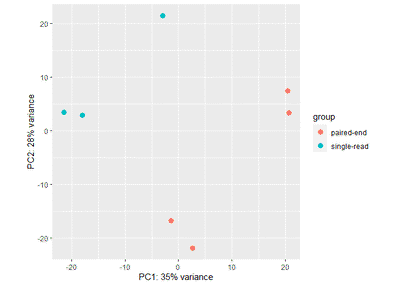
plotPCA(rld,intgroup=c("type"))
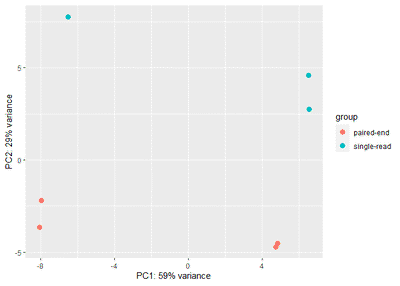
plotPCA(vsd,intgroup=c("type"))
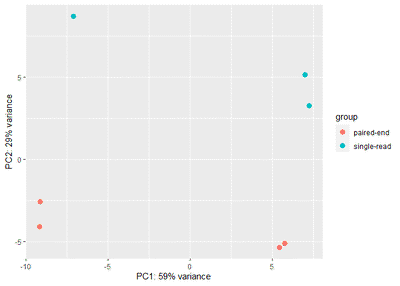
plot_PCA(t(norm),phe=input[[2]],group="type")
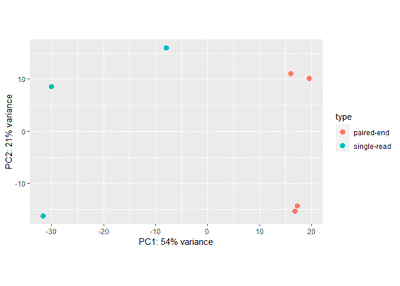
我们看到所有方法的规一化数据都表明,不同type之间的样本有明显的差别,即存在显著的批次效应。
4)矫正批次效应
1. 用limma去除批次效应
model <- model.matrix(~condition, colData(dds))
mat <- limma::removeBatchEffect(assay(ntd), ntd$type,design = model)
assay(ntd) <- mat
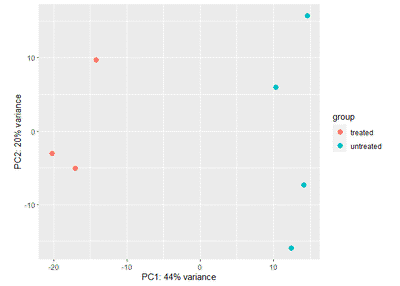
plotPCA(ntd,intgroup=c("condition"))
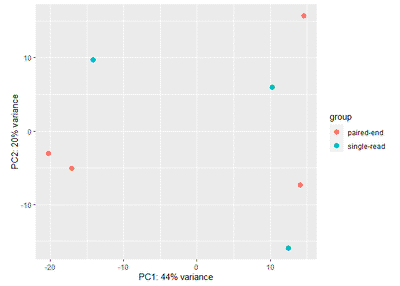
plotPCA(ntd,intgroup=c( "type"))
mat <- limma::removeBatchEffect(assay(rld), rld$type,design = model)
assay(rld) <- mat
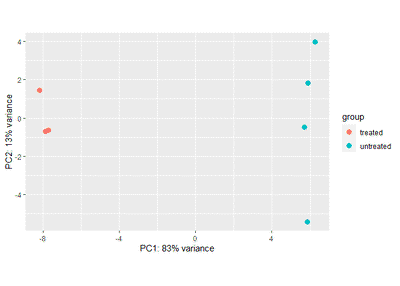
plotPCA(rld, intgroup=c("condition"))
plotPCA(rld, intgroup=c( "type"))
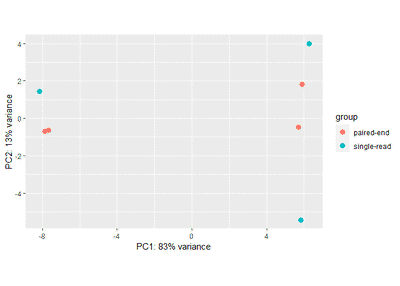
mat <- limma::removeBatchEffect(assay(vsd), vsd$type,design = model)
assay(vsd) <- mat
plotPCA(vsd, intgroup=c("condition"))
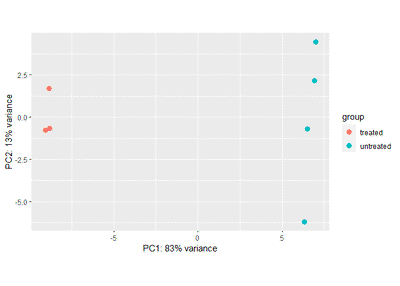
plotPCA(vsd, intgroup=c("type"))
可以看到样本可以被condition分开,但不能被type分开,表明矫正成功。
2. 用ComBat去除批次效应
library(sva)
model <- model.matrix(~condition,data = phe)
clean_norm <- norm[rowSums(norm)>1 & rowMedians(norm)>1,]
adjusted_expr <- ComBat(dat = clean_norm, batch = as.character(phe$type))
## Found 33 genes with uniform expression within a single batch (all zeros); these will not be adjusted for batch.
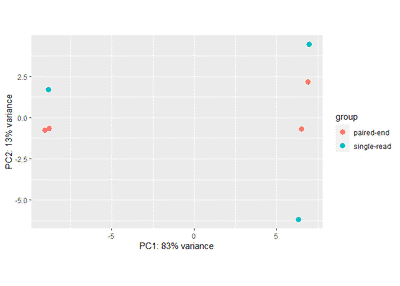
plot_PCA(t(adjusted_expr),phe=phe,group = "condition")
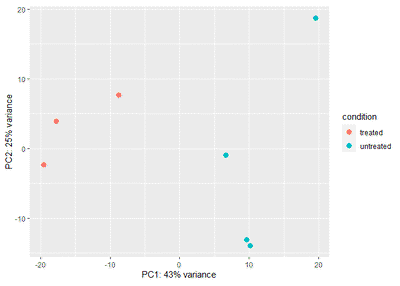
plot_PCA(t(adjusted_expr),phe=phe,group = "type")
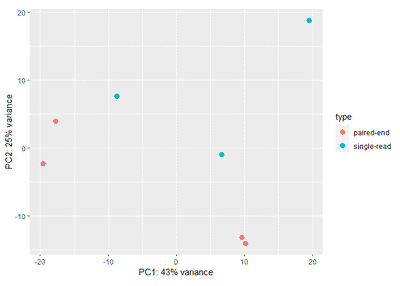
注意ComBat的输入文件是过滤且规一化的数据,不能是原始count。